

Fabry disease is a progressive disorder that starts in early childhood.
What is Fabry disease?
Fabry disease is an X-linked lysosomal storage disorder that affects men, women, and children of all ethnicities.1,2 The prevalence of Fabry disease is estimated to be 1:40,000 in males and 1:20,000 in females.1,3
Fabry is a progressive, multisystemic disorder that can result in potentially life-threatening damage to the kidney, heart, and brain.2,4
Early signs and symptoms of Fabry disease, which can start as early as childhood or adolescence, may include pain, gastrointestinal disturbances, angiokeratomas, hypohidrosis, proteinuria, and other signs and symptoms.5 As Fabry disease progresses, renal function deterioration and hypertrophic cardiomyopathy may occur, and the risk of developing fatal complications such as end-stage renal disease, stroke, cardiac fibrosis, arrhythmias, and premature death increases.1,5

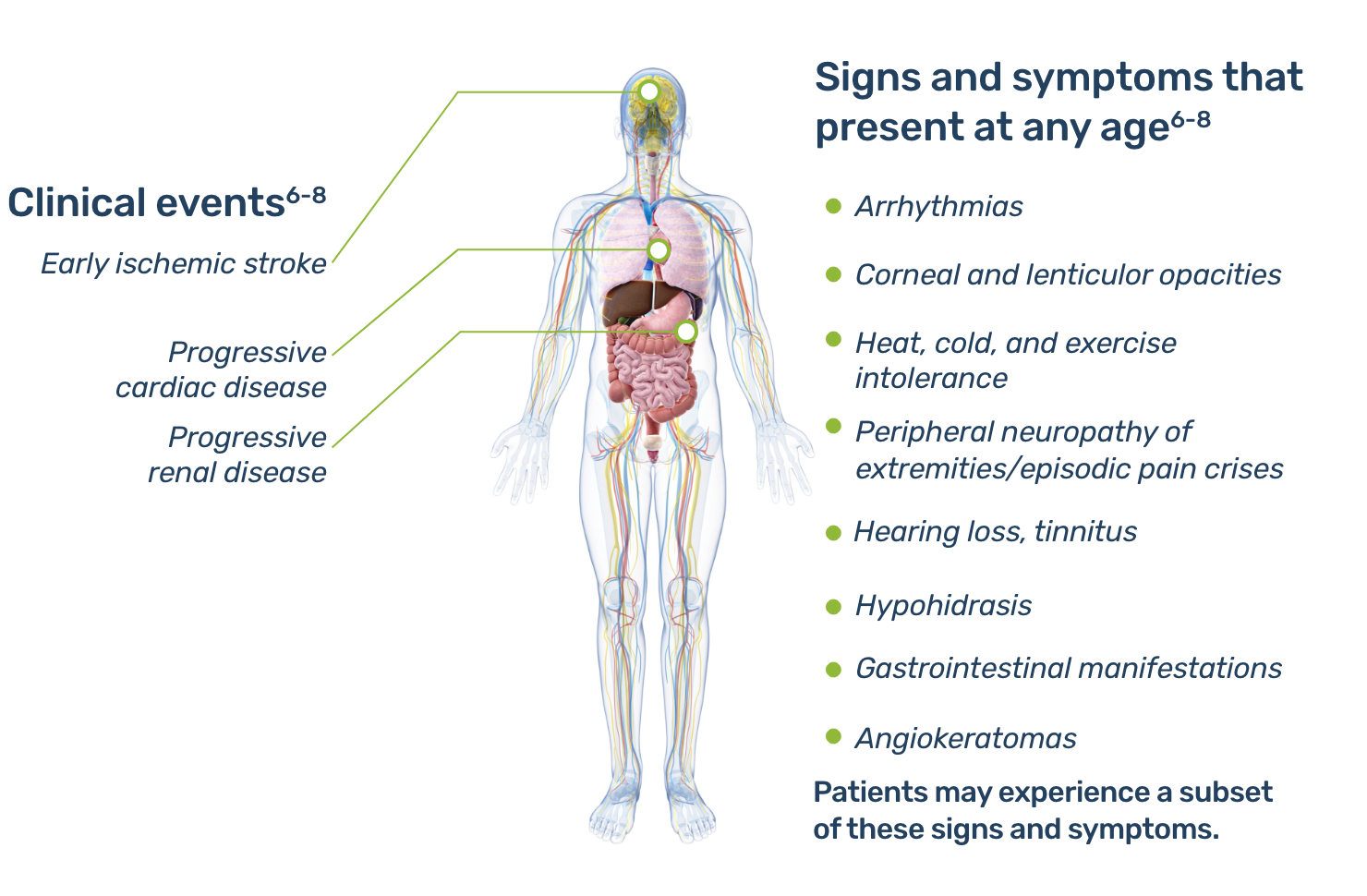
Learn more about manifestations of Fabry disease in your specialty.
Pathophysiology
In Fabry disease, variants in the GLA gene, located on the X chromosome, result in defects in the synthesis and/or function of alpha-galactosidase A (α-Gal A).9
α-Gal A is a lysosomal enzyme, which usually metabolizes globotriaosylceramide (also known as GL-3 or Gb3) and prevents it from accumulating.10
Complete or partial deficiency of α-Gal A leads to progressive accumulation of glycosphingolipids, particularly GL-3, in the lysosomes of numerous cell types.5
In Fabry disease, accumulation of GL-3 begins prenatally, or in utero, and continues over a lifetime.1,2 With advancing age, the progressive lysosomal GL-3 accumulation—particularly in the microvasculature—leads to renal failure, heart disease, stroke, and premature demise, typically in the fourth or fifth decade of life.4,5,10,11
The clinical manifestations of Fabry disease span a broad spectrum of severity and generally correlate with a patient's residual α-Gal A activity levels
Behind every patient diagnosed with Fabry is a family that is at risk.
For every index patient diagnosed, an average of 5 additional affected family members may be identified.

Overview of genetics
Over 900 variants in the GLA gene associated with Fabry disease have been identified.9,12 Notably, most families with classic disease have variants specific to their family.2
As an X-linked disease, the genetic defect that causes Fabry disease can be passed on by both males and females, and females are not simply carriers.1,5,13 Men have a 100% chance of passing the altered gene to their daughters and a 0% chance of passing it to their sons, while women with Fabry disease have a 50% chance of passing the altered gene to each daughter and son.5
Females with the altered GLA gene can develop Fabry, but the manifestations can be heterogeneous because of organ-and-tissue-specific X-inactivation.
Learn more about Fabry disease in women
Fabry disease is a genetic disorder; consequently, genetic counseling and family screening are important components of familial risk assessment.
Road to diagnosis
In a study based upon the Fabry Registry, due to the nonspecific and heterogeneous nature of early symptoms in Fabry disease, diagnostic delays and misdiagnosis were common.8

Fabry disease is often misdiagnosed and confused with rheumatoid or juvenile arthritis, rheumatic fever, erythromelalgia, Raynaud's syndrome, neurosis, lupus, acute appendicitis, or multiple sclerosis. Pain, particularly in children, can be dismissed as malingering.2,5
Connecting seemingly unrelated symptoms to Fabry disease can help avoid diagnostic delays and help patients receive disease management sooner.10
Importance of Screening for Fabry Disease in High-Risk Populations
Although Fabry is considered a rare disease, the prevalence of Fabry disease in patients with certain disorders, such as unexplained chronic kidney disease, hypertrophic cardiomyopathy, and premature stroke, is higher than in the general population.14-16 Consequently, it is important to screen patients with these conditions for Fabry disease.
Women with Fabry disease
In the past, female heterozygotes were erroneously described as "carriers of the defective gene" and believed not to develop symptoms of the disease. It is now known that if a female has a GLA variant, she is likely to become symptomatic over time.13 Moreover, her symptoms can be quite heterogeneous, ranging from the severe to the less common, seemingly asymptomatic disease course. This range of heterogeneity in symptomology makes diagnosis of Fabry in females particularly challenging.
Once diagnosed, it is important to monitor females who appear asymptomatic for symptom development.
According to the Registry, 69.4% of women with Fabry disease develop symptoms related to Fabry disease over time.8
This variability in presentation of Fabry disease in females is due to patterns of X-inactivation. Females have two X chromosomes, and genes on one X chromosome are randomly inactivated. Each organ in a female's body has its own X-inactivation pattern. The resulting organ- and tissue-specific X-inactivation of GLA in females leads to corresponding variations in organ-and tissue-specific expression of α-Gal A, which cause heterogeneous manifestations and presentation.13
Females can have Fabry disease despite having what appears to be normal α-Gal A activity in plasma. In a cross-sectional study of 57 symptomatic heterozygous women, some women with normal enzyme activity had cardiac, renal, or cerebrovascular abnormalities.17
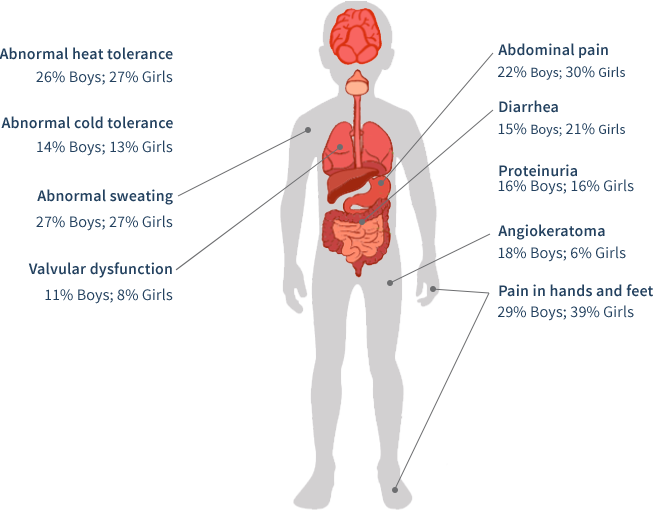
Organ System Involvement in Females With Fabry Disease4

Signs and symptoms by life stage
The age of presentation of Fabry disease is variable, as are the presenting symptoms and the clinical course. A subset of disease symptoms usually appear during childhood.18 However, the disease often goes unrecognized until adulthood, when more severe symptoms prompt patients to pursue diagnosis. Earlier diagnosis may result in earlier disease management.
Fabry disease in your practice
Patients with Fabry disease often have a variety of symptoms and clinical manifestations across multiple organ systems. Consequently, patients are often referred to specialists for further evaluation, and specialists may be first to raise suspicion of and ultimately diagnose Fabry disease. Below is an overview of how Fabry disease may present in your practice.
Kidney disease is a major complication of Fabry Disease.10,20 The prevalence of Fabry disease in the dialysis population is approximately 100 to 1000 times higher than in a reference population.14-16,21,22 Patients with Fabry disease are at high risk of progressing to end-stage renal disease (ESRD) at a young age (typically 30s to 50s but ESRD has been reported as early as teen years).20 The prevalence of chronic kidney disease (CKD) in patients with Fabry disease is approximately 5 times higher than in the general population.8,23
Renal damage as a result of GL-3 accumulation in various renal cells can start as early as the first decade of life, often preceding laboratory abnormalities and clinical symptom onset.20,24
In males and females, progressive accumulation of GL-3 in podocytes, followed by podocyte injury, manifests later as proteinuria and reduced glomerular filtration rate, which can ultimately lead to CKD or progression to end-stage renal disease (ESRD).25,26
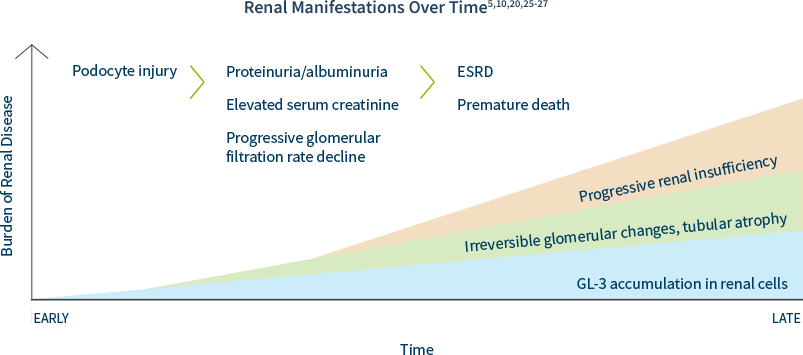
Including Fabry disease in the differential diagnosis of unexplained ESRD is key to identifying patients and families.
European Renal Best Practice recommends screening males under age 50 with unexplained CKD and females of any age with unexplained CKD and other symptoms associated with Fabry disease.28
If you have a patient that you suspect may have Fabry disease, consider having them and their family tested.
Cardiac disease is the most common cause of death in patients with Fabry disease.24 Unexplained cardiac manifestations could indicate Fabry disease. The prevalence of Fabry disease in patients with left ventricular hypertrophy (LVH) or hypertrophic cardiomyopathy (HCM) is estimated to be at least 1 in 100.29-31
Forty-nine percent of males and 35% of females with the disease were found to have had a cardiac event by an average age of 36 and 44, respectively—and cardiac events may appear as early as the teen years.27
Cardiac hypertrophy, fibrosis, and conduction abnormalities can be caused by GL-3 accumulation in cellular components of the heart, adversely affecting cardiac structure and function. Other cardiac manifestations may include: EKG abnormalities, short PR intervals, AV block, repolarization abnormalities, ST-T changes, and/or arrhythmias.32 Persistent accumulation of GL-3 in cardiomyocytes over time may lead to LVH, ultimately resulting in heart failure.33

Including Fabry disease in the differential diagnosis of unexplained LVH or HCM is key to identifying patients and families.
The European Society of Cardiology guidelines recommend a systematic search for the underlying cause of increased LV wall thickness, including specialized laboratory testing and genetic analysis.38 Screening patients with unexplained LVH or HCM for Fabry disease is key to identifying patients and families.
If you have a patient that you suspect may have Fabry disease, consider having them and their family tested.
Prevalence estimates of Fabry disease in patients with premature stroke range from 1% to nearly 5%, much higher than in the general population.39
Based on registry data, the median age of the first stroke is 39 years in men and 46 years in women.5
In Fabry disease, GL-3 can accumulate in peripheral and central neurons as well as cerebral blood vessels, causing:39,40
• Neuropathic pain and autonomic symptoms, due to damage to peripheral neurons and axons
• Cognitive impairment and mood disorders, as evidenced by white matter lesions
PNS: Neuropathic Pain
GL-3 accumulation causes neural damage primarily involving the small nerve fibers of the peripheral somatic and autonomic nerve systems. Pain is one of the earliest symptoms of Fabry, affecting 60% to 80% of classically affected boys and girls, which affects their quality of life. Boys generally have an earlier age of related symptom onset than girls. Two types of pain have been described:5
• Episodic crises ("Fabry crises")—agonizing burning pain originating in the extremities
and radiating inward to the limbs and other parts of the body
• Chronic pain—burning and tingling paraesthesia
CNS Complications
The early peripheral neuropathic hallmarks of Fabry disease are often followed by cerebrovascular complications and autonomic dysfunction in adulthood.5
Fabry disease is often misdiagnosed as multiple sclerosis (MS) due to white matter lesions.39,41
Some of the most devastating neurological features of Fabry disease are caused by cerebrovascular lesions, which are the result of multifocal involvement of small blood vessels. Cerebrovascular involvement can lead to a wide variety of signs and symptoms ranging from mild to severe, including headache, vertigo/dizziness, transient ischemic attacks, and ischemic strokes.5
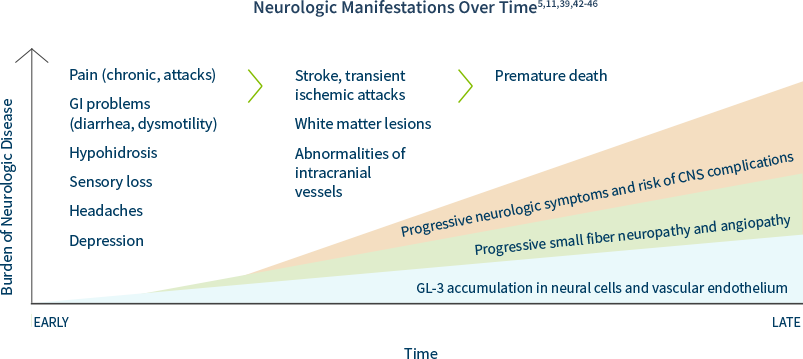
If you have a patient that you suspect may have Fabry disease, consider having them and their family tested.
GL-3 and other lipids may deposit in the eyes of Fabry disease patients, resulting in a wide range of ocular findings and/or visual complaints. These findings typically do not affect the patient's vision but may be pathognomonic for Fabry disease.2,47
One of the earliest and most common signs of Fabry disease is corneal whorling, seen in 80% of patients with Fabry disease. The earliest report in boys is age 2 and in girls is age 6. Corneal whorling can be detected with a routine slit lamp examination.47

Distinctive Corneal Verticillata in Fabry 2,31,47
Corneal whorling is a symmetric, bilateral, whorl-like pattern of powdery, white, yellow, or brown corneal epithelial GL-3 deposits emanating from a single vortex.47
A number of medications, such as amiodarone and chloroquine, can also cause this phenomenon. Patients with Fabry disease may suffer from arrhythmias; therefore, Fabry should not be ruled out in a patient found to be on amiodarone.
In addition to corneal opacities, patients with Fabry disease may present with47:
• Posterior capsular cataracts with whitish spoke-like deposits of granular material (Fabry cataract)
• Aneurysmal dilatation of thin-walled venules on the bulbar conjunctiva
• Mild-to-marked tortuosity and angulation of the retinal vessels
The most recognizable clinical feature of Fabry disease is angiokeratomas, which have the following characteristics1,2:
• Dark red or purple skin lesions
• Range in size from a pinpoint to several millimeters in diameter
• Do not blanch with pressure
• Are usually distributed on the buttocks, groin, umbilicus, and upper thighs (bathing suit distribution)

Angiokeratomas in Fabry disease1,2
Characteristic dark red to blue-black angiectases are typically found between the thigh (left) and umbilicus (right) regions ("bathing suit distribution"). Used with permission from R.J. Desnick, MD, PhD.
Lesions generally appear in adolescence or young adulthood and may become larger and more numerous with age.2 Angiokeratomas are almost universal in male hemizygotes; they occur in approximately 30% of heterozygous females.1
Accumulation of GL-3 may begin prenatally or in utero and continue over a lifetime. Clinical onset of Fabry disease occurs in childhood, and presentation may be subtle. Signs and symptoms are often discounted as malingering or are mistakenly attributed to other disorders, such as rheumatic fever, erythromelalgia, neurosis, Raynaud's syndrome, multiple sclerosis, chronic intermittent demyelinating polyneuropathy, lupus, acute appendicitis,"growing pains," or petechiae.2
The early clinical course of Fabry disease typically involves signs and symptoms that primarily affect quality of life: chronic pain, angiokeratomas, hypohidrosis, heat and cold intolerance, and gastrointestinal symptoms.2,19
Pain is one of the earliest symptoms of Fabry, affecting 60% to 80% of classically affected boys and girls, which affects their quality of life. Boys generally have an earlier age of related symptom onset than girls.5
An often overlooked manifestation of Fabry disease is the presence of GI symptoms, including abdominal pain (often after eating), diarrhea, nausea, and vomiting, which are a significant cause of anorexia. Diarrhea-predominant irritable bowel syndrome (IBS) is a differential diagnosis.5
In a pediatric study based upon the Fabry Registry, boys and girls with Fabry disease begin developing symptoms at an early age (median age of 6 years for boys [n=194] and girls [n=158]).48 Moreover, some may have early severe complications.48
Primary care professionals (PCPs) often have the most frequent contact with patients. PCPs are often In the best position to first observe the cluster of seemingly unrelated signs and symptoms associated with Fabry disease. PCPs can help end a long patient journey in a proper diagnosis. Consequently, it is important they be aware of the common presenting symptoms for Fabry disease.
Common Presenting Signs and Symptoms for Fabry 2,5,19
• Fatigue
• Gastrointestinal problems such as abdominal pain, diarrhea, vomiting, and nausea
• Heat and cold intolerance
• Decreased ability to sweat (hypohidrosIs/anhidrosis)
• Neuropathic pain in the hands and feet, or acute agonizing episodes of radiating pain
in the extremities or abdomen ("Fabry crises”) lasting for minutes or days
• Recurrent fever accompanying pain and associated with elevated erythrocyte sedimentation rate
• A more specific sign of Fabry disease that may be seen in adolescents and adults is the presence
of angiokeratomas—dark reddish-purple skin lesions that do not blanch with pressure
• Depression
These presenting symptoms in conjunction with a family history of early renal or cardiac disease should raise a high suspicion of Fabry disease.
Gastrointestinal (GI) symptoms of Fabry disease may be related to the deposition of GL-3 in the autonomic ganglia of the bowel and mesenteric blood vessels.5
Despite being common, GI symptoms remain an under recognized manifestation of Fabry. Up to two-thirds of affected males and about half of symptomatic females may experience GI symptoms associated with Fabry disease.5,49,50 GI symptoms often appear In childhood and usually remain present during adulthood.
Patients may complain of abdominal pain (often after eating), diarrhea, nausea, and vomiting, which are a significant cause of anorexia. Diarrhea-predominant irritable bowel syndrome (IBS) is a differential diagnosis.5
Fatigue and pain may masquerade as rheumatological conditions such as lupus, chronic fatigue syndrome, and rheumatoid or juvenile arthritis.3 In a rheumatology setting, the most common presenting symptoms of Fabry disease are1,2,5,51:
• Joint pain
• Acute and chronic hand or foot pain, or acute agonizing episodes of radiating pain In the
extremities lasting minutes to days ("Fabry crises")
• Angiokeratomas: dark reddish-purple skin lesions that do not blanch with pressure and
may resemble vasculitis
• Recurrent fever accompanying pain and associated with elevated erythrocyte sedimentation rate
Find more information about Fabry disease
- Connect with a Sanofi Representative
- Information about Fabry disease
1. Desnick RJ, et al. In: The Online Metabolic and Molecular Bases of Inherited Diseases. New York, NY: McGraw Hill; 2014:1-64. 2. Desnick RJ. Ann Intern Med. 2003;138(4):338-346. 3. Laney DA, et al. J Genet Couns. 2008;17(1):79-83. 4. Eng CM, et al. J Inherit Metab Dis. 2007;30(2):184-192. 5. Germain DP. Orphanet J Rare Dis. 2010;5(30):1-49. 6. Barba-Romero MA, et al. Int J Clin Pract. 2011;65(8):903-910. 7. Mehta A. QJM. 2002;95(10):647-653. 8. Wilcox WR, et al. Mol Genet Metab. 2008;93(2):112-128. 9. Nagueh SF. Circulation. 2014;130(13):1081-1090. 10. Ortiz A, et al. Nephrol Dial Transplant. 2008;23(5):1600-1607. 11. Sims K. et al. Stroke. 2009:40(3):788-794. 12. The Human Gene Mutation Database. Institute of Medical Genetics in Cardiff. Available at: http://www.hgmd.cf.ac.uk/ac/index.php. Accessed January 19, 2018. 13. Wang RY et al. Genet Med 2007;9(1):34-45. 14. Nakao S, et al. Kidney Int. 2003;64(3):801.807. 15. Linthorst GE, et al. Nephrol Dial Transplant. 2003;18(8):1581-1584. 16. Kotanako P, et al. J Am Soc Nephrol. 2004;15(5):1323-1329. 17. Gupta S, et al. Medicine (Baltimore). 2005;84(5):261-268. 18. Shelley ED, et al. Pediatr Dermatol 1995;12(3):215-219. 19. Fabry Registry Annual Report 2010. Available at: www.fabry.org/fsig.nsf/PDFs/PDFsR/$File/2010_Annual_Report.pdf. Accessed January 19. 2018. 20. Ramaswami U, et al. Clin J Am Soc Nephrol. 2010;5(2):365-370. 21. Ichinose M, et al. Clin Exp Nephrol. 2005;9(3):228-232. 22. Bekri S, et al. Nephrol Clin Pract 2005:101(1):c33-38. 23. Coresh J, et al. JAMA. 2007;298(3):2038-2047. 24. Waldek S, et al. BMC Nephrol. 2014:15(72):1-15. 25. Najafian B, et al. Kidney Int. 2011;79(6):663.670. 26. Torra R. Kidney Int. Suppl. 2008;(Suppl. 111):S29-S32. 27. Schiffmann R, et al. Nephrol Dial Transplant. 2009.24(7) 2102-2111. 28. Terryn W, et al. Nephrol Dial Transplant. 2013;28(3):505.517. 29. Linthorst GE, et al. J Med Genet. 2010;47(4):217-222. 30. Monserrat L, et al. Coll Cardiol. 2007;50(25):2399-2403. 31. van der Tol L, et al. J Med Genet. 2014;51(1):1-9. 32. Yousef Z, et al. Eur Heart J. 2013;34(11):802-808. 33. Eng CM, et al. Genet Med. 2006;8(9):539-548. 34. Linhart A, et al. Eur Heart J. 2007;28(10):1228-1235. 35. Patel MR, et al. J Am Coll Cardiol. 2011;57(9):1093-1099. 36. Kampmann C, et al. Int J Cardiol. 2008:130(3):367-373. 37. Weidemann F, et al. Orphanet J Rare Dis. 2013:8(116). 38. Elliott PM, et al. Eur Heart J. 2014;35(39):2733-2779. 39. Fellgiebel A, et al. Lancet Neurol. 2006;5(9):791-795. 40. Hilz MJ. Clin Ther. 2010;32 (suppl C):S93. 41. Bottcher T. et al. Plos ONE. 2013:8(8):e71894. 42. Cable WJL, et al. Neurology. 1982;32(5):498-502. 43. Uceyler N, et al. Clin J Pain. 2014;30(10):915-920. 44. Fellgiebel A, et al. Neurology. 2009;72(1):63-68. 45. Moore DF, et al. Brain Res Bull. 2003;62(3):231-240. 46. Cole AL, et al. J Inherit Metab Dis. 2007:30(6):943-951. 47. Samiy N, et al. Surv Ophthalmol. 2008;53(4):416-423. 48. Hopkin RJ, et al. Pediatr Res. 2008;64(5):550-555. 49. MacDermot KD, et al. J Med Genet. 2001a;38(11):750-760. 50. MacDermot KD, et al. J Med Genet. 2001b;38(11):769-775. 51. Manger B, et al. Clin Rheumatol. 2007;26(3):335-341.